



A bit of bioinformatics

Context
10 to 100 million species live on earth.
Evolution is the source of this diversity: all species are related, to varying degrees. Within each species, individuals evolve over generations through random changes in their DNA.
These changes (mutations) will be selected, according to the environment and the interaction of species and individuals within them. Sometimes, a new characteristic or even a new species appears, which is better adapted to its environment.
In order to define these relationships, biologists look not only at what species have in common, but also what distinguishes them. The relationships between different species are most often represented in the form of a tree, called a ‘phylogenetic tree’ or ‘tree of life’.
In Darwin’s time, species were first compared on the basis of their morphology – for example, analyses of the size, shape and structure of bones, the presence of hair or scales or, for plants, the position of leaves on a stem. The more similar the morphological characteristics of two species are, the more recent their common ancestor is.
Today, it is possible to study the evolution of species by comparing their DNA and in particular, their genes or proteins. The more similar the DNA of two species is, the more recent their common ancestor is.
What is bioinformatics?
What is bioinformatics? How to sequence and annotate a genome? How to predict genes? How to find orthologs?
The challenges of building a tree of life with molecular data are multiple: among other things, it is necessary to have data that are comparable, such as sets of orthologous protein sequences. And these data are obtained from the genome sequences of different species! This is where bioinformatics comes into play!
How to sequence a genome?
1. Sequencing
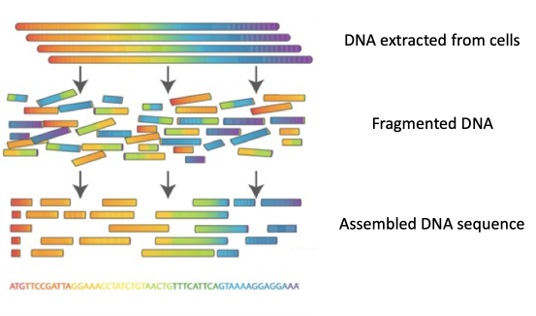
Thanks to new technologies, it is now possible to sequence entire genomes (‘whole genome sequencing (WGS)’), that is to determine the order in which the 4 nucleotides A, C, T, G are found in the DNA of all chromosomes.
The steps: DNA is extracted from the cells of an organism, and then cut into tiny pieces (fragmented) in a random manner. Each fragment is copied several hundred of times before being sequenced using a machine known as a DNA sequencer.
Presently, more than 10 billion DNA fragments with a length ranging from 150 to 300 nucleotides can be sequenced in just a few hours! The DNA sequences obtained in this manner are then stored on a computer.
More info
2. Assembling
The billions of DNA fragments (called ‘reads’) are then assembled, a bit like the pieces of a puzzle, to reconstitute the sequence of each chromosome.
Many algorithms are able to assemble genomes de novo, i.e. without the help of a reference genome to rely on – a bit like a puzzle that needs to be put together without having the final picture.
Your tum to play: assemble the DNA fragments!
Print the DNA fragments and cut them into strips of paper. These DNA fragments can be assembled, as they overlap. Find the DNA sequence of the genome!
>sequence1 tggtctagctacagtgaaatctcgatgg >sequence2 agctacagtgaaatctcgatgg >sequence3 ggtgattttggtctag >sequence4 ggtgattttggtctagctacagtg >sequence5 ggtgattttggtctagctacag >sequence6 tacagtgaaatctcgatgg >sequence7 ggtgattttggtctagctacagt >sequence8 ttttggtctagctacagtgaaatctcgatgg >sequence9 ggtgattttggtctagctacagt

3. Annotating
Once the sequence of the genome is known, the next step consists of finding where the genes are located in this immense string of characters…and to predict their biological function. This is what is referred to as ‘annotating the genome’.
Things are not easy…

How to predict the genes?
Gene lengths are very variable, ranging between 10,000 and 10 million nucleotides, and are embedded in the genome. The genes coding for proteins in human only constitute a small part – less than 5% – of our DNA. That’s like looking for a needle in a haystack!
A file containing the sequence of a 3 billion nucleotides genome (the human genome, for example) would take up 3 Gb of computer disk space! To give you an idea of the magnitude of the task, here is just the DNA sequence of human chromosome 1…
This is why scientists have imagined and written bioinformatics programs capable of predicting the beginning of a gene, its end, what is in between, and the corresponding protein.
These programs look for, among other things, short nucleotide sequences known to be present in the genes (in green). While these programs still do not return perfect results, they provide biologists with precious information.
Indeed, there are now far too many sequenced genomes for all these data to be validated experimentally!

The detective work is not over! Bioinformaticians still need to look for orthologs (mainly orthologous proteins) so they can start comparing them to study genome similarities and differences… and thus learn more about evolution.
The bioinformatics quest for orthologs
The human insulin protein is the ortholog of the macaque insulin protein or of the bovine insulin protein. These three proteins are derived from a common ancestral gene. But how to know it?
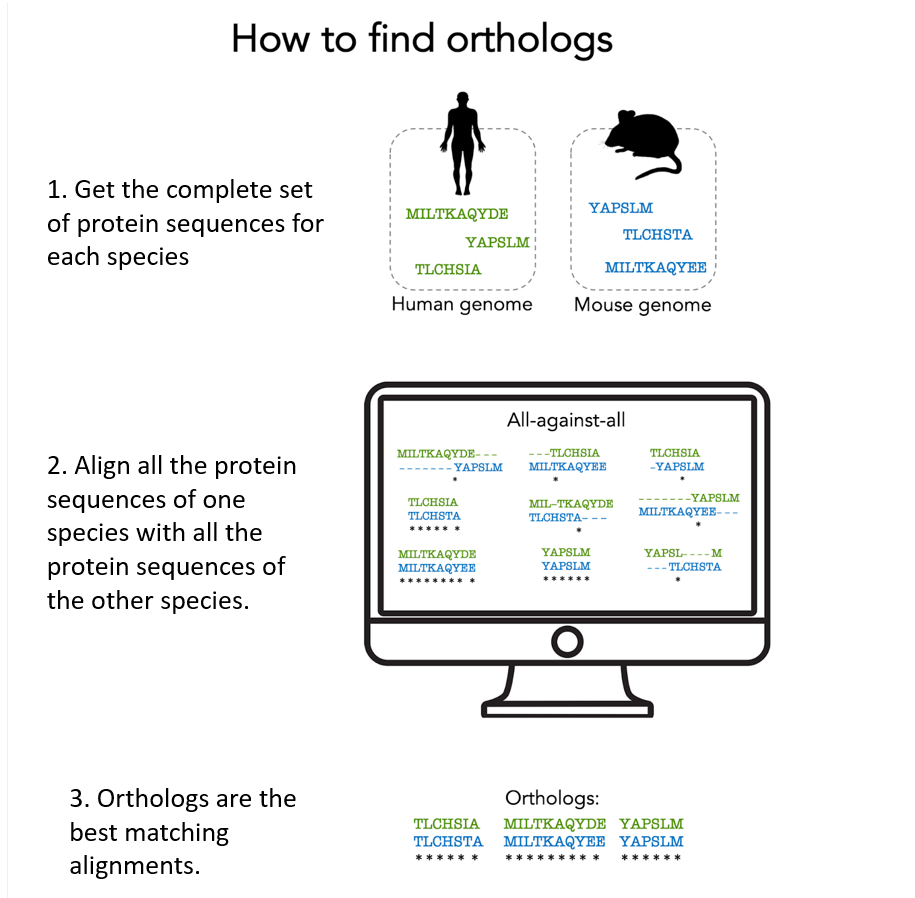
To find orthologous proteins, all proteins from one species must be compared to all proteins from another species to find which ones ‘match’, i.e. are derived from a common ancestral gene.
Here are the main steps of this ‘quest for orthologs’:
1. Get all protein sequences produced by a given organism.
These protein sequences are available in the UniProtKB database, for example. UniProtKB stores some 220 million protein sequences from over 750,000 organisms.
2. Align 2 by 2 all the proteins of one organism with those of another. There are a lot of alignments to do! We need powerful computers to do all these calculations.
3. Orthologs are derived from a single ancestral gene present in the common ancestor of the species in concern: they are typically identified as the protein pairs with the best alignment score. The computer program takes into account all biological processes that may have occurred (gene duplication, gene loss, …).
Once the list of orthologs is established, we can start looking for similarities and differences between species to build trees to study evolution!
