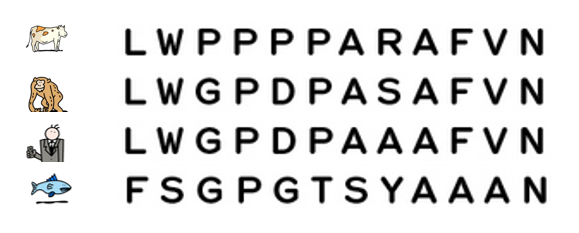Context
10 to 100 million species live on earth.
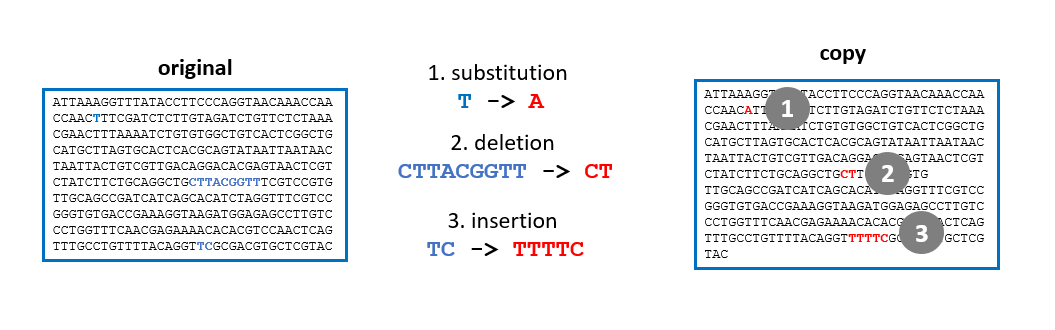
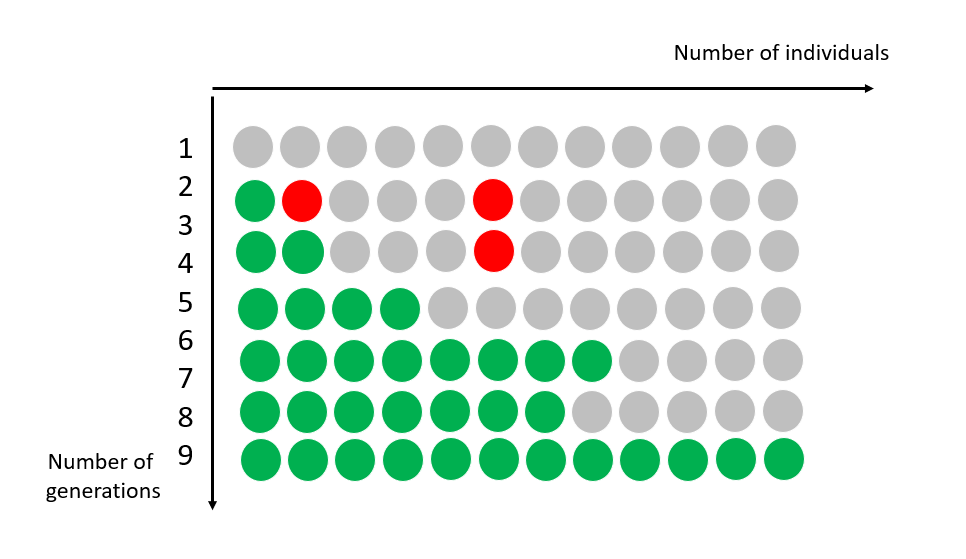
Evolution is the source of this diversity: all species are related, to varying degrees. Within each species, individuals evolve over generations through random changes in their DNA.
These changes (mutations) will be selected, according to the environment and the interaction of species and individuals within them. Sometimes, a new characteristic or even a new species appears, which is better adapted to its environment.
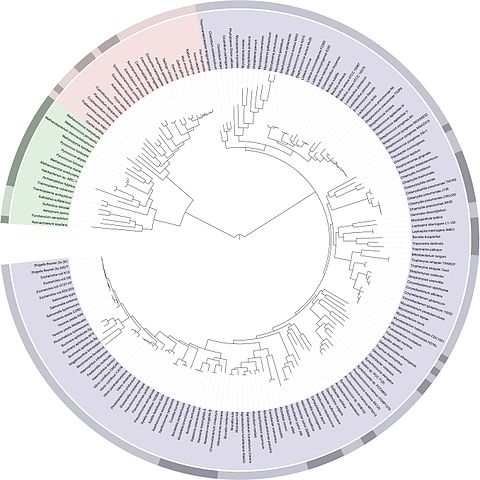
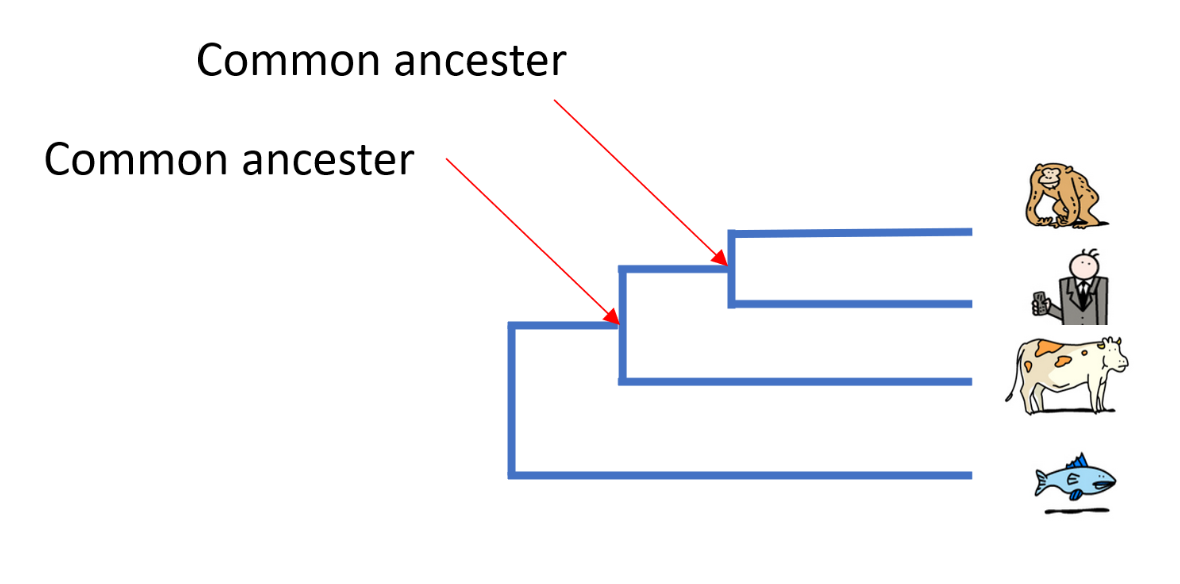
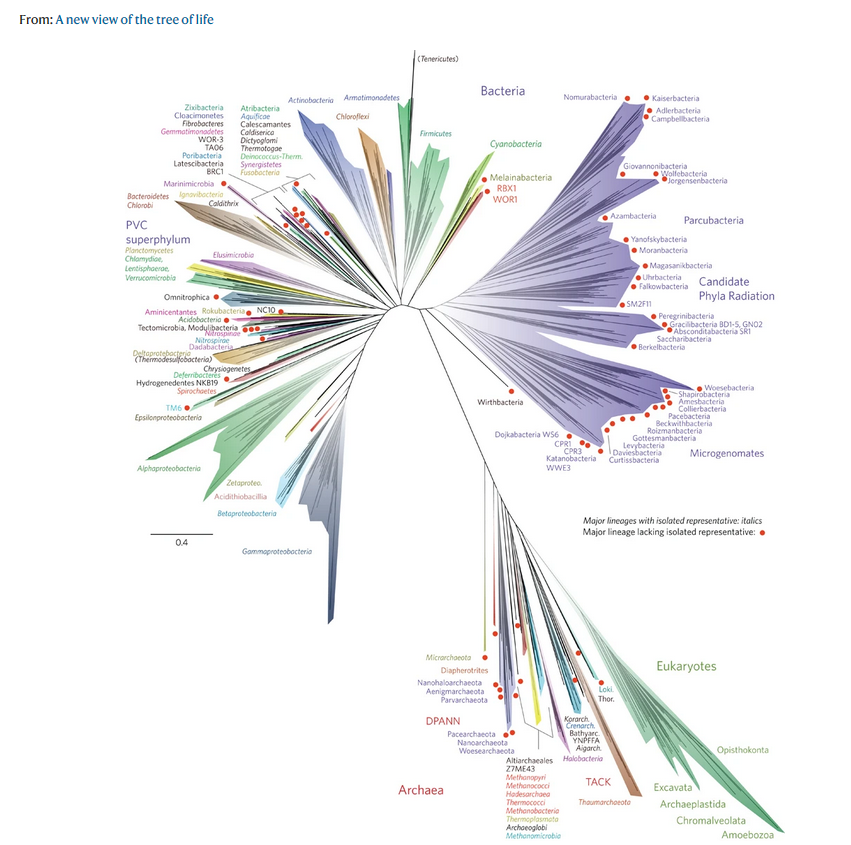
In order to define these relationships, biologists look not only at what species have in common, but also what distinguishes them. The relationships between different species are most often represented in the form of a tree, called a ‘phylogenetic tree’ or ‘tree of life’.
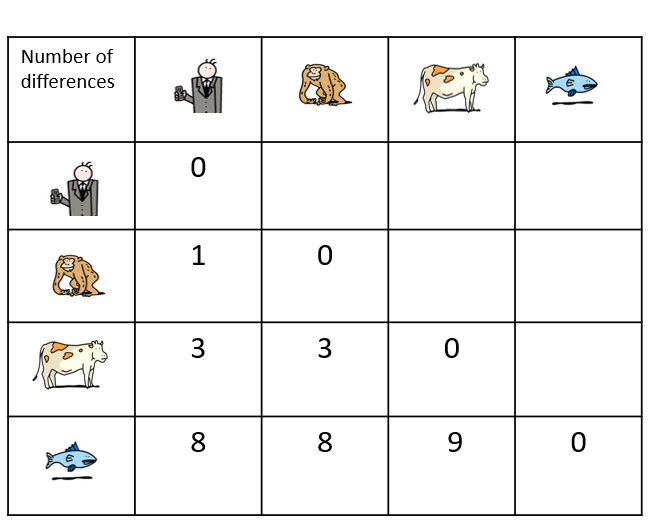
In Darwin’s time, species were first compared on the basis of their morphology – for example, analyses of the size, shape and structure of bones, the presence of hair or scales or, for plants, the position of leaves on a stem. The more similar the morphological characteristics of two species are, the more recent their common ancestor is.
Today, it is possible to study the evolution of species by comparing their DNA and in particular, their genes or proteins. The more similar the DNA of two species is, the more recent their common ancestor is.