



Un peu de bioinformatique

Le contexte
10 à 100 millions d’espèces peuplent notre planète.
L’évolution est à l’origine de cette diversité: toutes les espèces sont apparentées à des degrés divers. Au sein de chaque espèce, les individus évoluent au fil des générations grâce à des changements qui surviennent de façon aléatoire dans leur ADN.
Ces changements (mutations) vont être sélectionnés, a posteriori, en fonction de l’environnement et de l’interaction des espèces et des individus entre eux. Et parfois, une nouvelle caractéristique voire une nouvelle espèce apparaît, mieux adaptée à son environnement.
Pour définir les liens de parenté, les biologistes recherchent ce que les espèces ont en commun mais aussi ce qui les distingue. Les liens de parenté entre les différentes espèces sont représentés le plus souvent sous forme d’un arbre, appelé ‘arbre phylogénétique’ ou ‘arbre de la vie’.
A l’époque de Darwin, on commença par comparer les espèces sur la base de leur morphologie – analyses de la taille, de la forme et de la structure des os, la présence de poils ou d’écailles ou, pour les plantes, la position des feuilles sur une tige, par exemple. Plus les caractéristiques morphologiques de 2 espèces sont similaires, plus leur ancêtre commun est récent.
Aujourd’hui, il est possible d’étudier l’évolution des espèces en comparant leur ADN et en particulier, leurs gènes ou leurs protéines. Plus l’ADN de 2 espèces est similaire, plus leur ancêtre commun est récent.
C’est quoi la bioinformatique ?
C'est quoi la bioinformatique ? Comment séquencer un génome ? Comment prédire les gènes ? Comment trouver les orthologues ?
Les défis pour construire un arbre de la vie avec les données moléculaires sont multiples: il faut entre autre avoir accès à des données qui soient comparables, comme par exemple des sets de séquences de protéines orthologues. Et ces données sont obtenues à partir des génomes des différents organismes! C’est là que la bioinformatique entre en jeu!
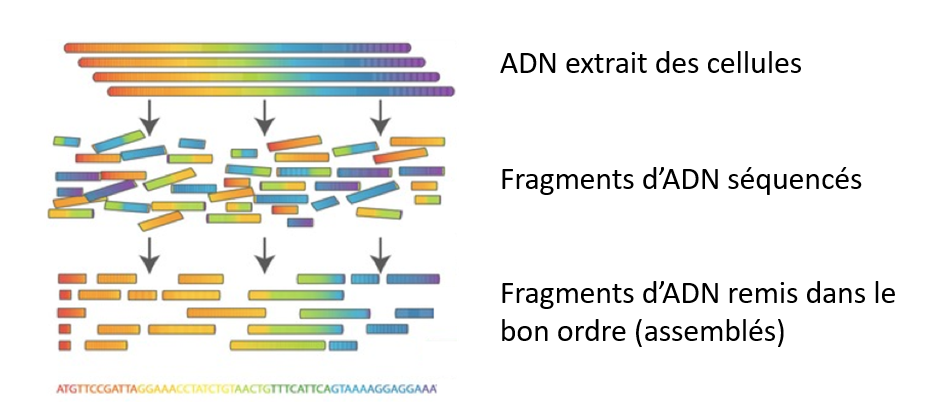
1. Séquencer
Grâce aux nouvelles technologies, il est désormais possible de séquencer des génomes entiers (‘whole genome sequencing (WGS)’), c’est-à-dire de déterminer l’ordre dans lequel se succèdent les 4 nucléotides A, C, T, G dans un des 2 brins de l’ADN en quelques heures.
Les étapes: l’ADN est extrait des cellules d’un organisme, il est ensuite fragmenté de façon aléatoire. Chaque fragment est copié plusieurs centaines de fois avant d’être séquencé à l’aide de machines appelées séquenceurs.
Aujourd’hui, plus de 10 milliards de fragments d’ADN d’une longueur de 150 à 300 nucléotides peuvent être séquencés en quelques heures! Les séquences ADN ainsi obtenues sont ensuite stockées dans un ordinateur.
Comment séquencer un génome ?
2. Assembler
Les milliards de fragments d’ADN (appelés ‘reads’) sont ensuite assemblés, un peu comme les pièces d’un puzzle, pour reconstituer la séquence de chaque chromosome.
Un grand nombre d’algorithmes sont capables d’assembler des génomes de novo, c’est-à-dire sans l’aide d’un génome de référence sur lequel s’appuyer – un peu comme un puzzle qu’il faudrait assembler sans avoir l’image finale.
A vous de jouer: assembler des fragments d'ADN !
Amusez-vous à assembler ces 9 fragments d’ADN, imprimés sur des bandelettes de papier et trouver la séquence ADN du génome. Ces fragments d’ADN peuvent être assemblés, car ils se chevauchent.
>sequence1
tggtctagctacagtgaaatctcgatgg
>sequence2
agctacagtgaaatctcgatgg
>sequence3
ggtgattttggtctag
>sequence4
ggtgattttggtctagctacagtg
>sequence5
ggtgattttggtctagctacag
>sequence6
tacagtgaaatctcgatgg
>sequence7
ggtgattttggtctagctacagt
>sequence8
ttttggtctagctacagtgaaatctcgatgg
>sequence9
ggtgattttggtctagctacagt

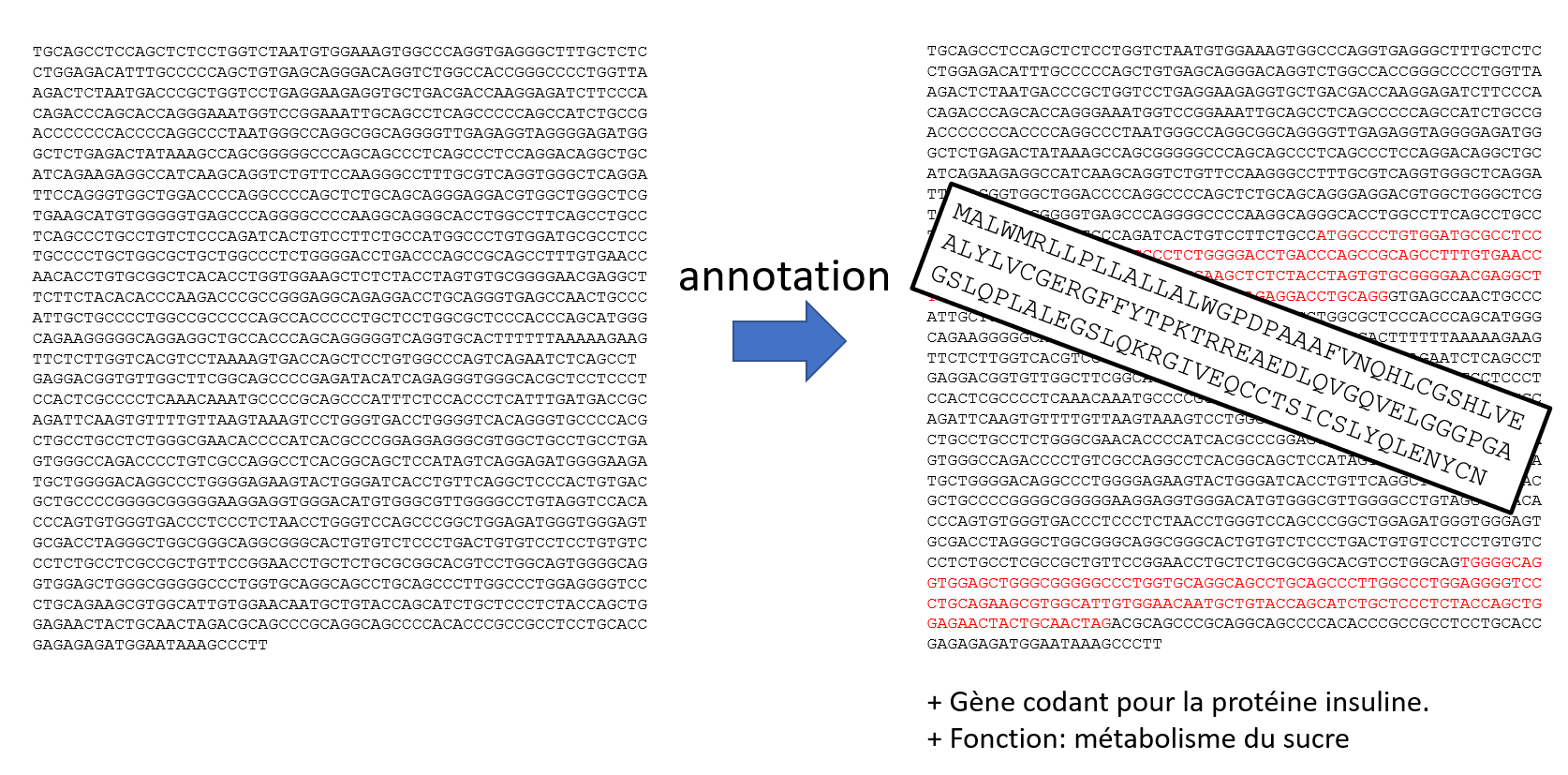
3. Annoter
Une fois la séquence du génome connue, l’étape suivante consiste à trouver où sont localisés les gènes dans cet immense texte…et de prédire leur fonction biologique. C’est ce que l’on appelle ‘annoter le génome’.
Les choses ne sont pas simples…
Comment prédire les gènes ?
Les gènes ont des longueurs très variables, comprises entre 1’000 et 10 millions de nucléotides et sont dispersés dans le génome. Les gènes qui codent pour des protéines chez l’homme ne constituent qu’une partie minime – moins de 5 % – de notre ADN. Autant chercher une aiguille dans une botte de foin!
Un fichier contenant la séquence d’un génome de 3 milliards de nucléotides (le génome humain par exemple) occuperait 3 Go d’espace disque !
Pour vous donner une idée l’ampleur de la tâche, voici juste la séquence ADN du chromosome 1 humain…
C’est pourquoi on a imaginé et conçu des programmes bioinformatiques capables de prédire le début et la fin d’un gène, les exons et la séquence de la protéine correspondante.
Ces logiciels cherchent, entre autres, des petites séquences nucléotidiques connues pour être présentes dans les gènes (en vert). Ces programmes sont encore loin de donner des résultats parfaits mais offrent de précieuses indications aux biologistes. En effet, il y a aujourd’hui beaucoup trop de génomes séquencés pour que toutes ces données puissent être validées expérimentalement !

Le travail de détective n’est pas terminé! Les bioinformaticiens doivent encore chercher les orthologues (principalement les protéines orthologues) afin de pouvoir commencer à les comparer pour étudier les similitudes et les différences… et en apprendre ainsi davantage sur l’évolution.
Comment trouver les orthologues ?
La protéine insuline de l’homme est l’orthologue de la protéine insuline du macaque ou de la protéine insuline du boeuf . Ces 3 protéines ont un gène ancestral commun. Mais comment le savoir ?
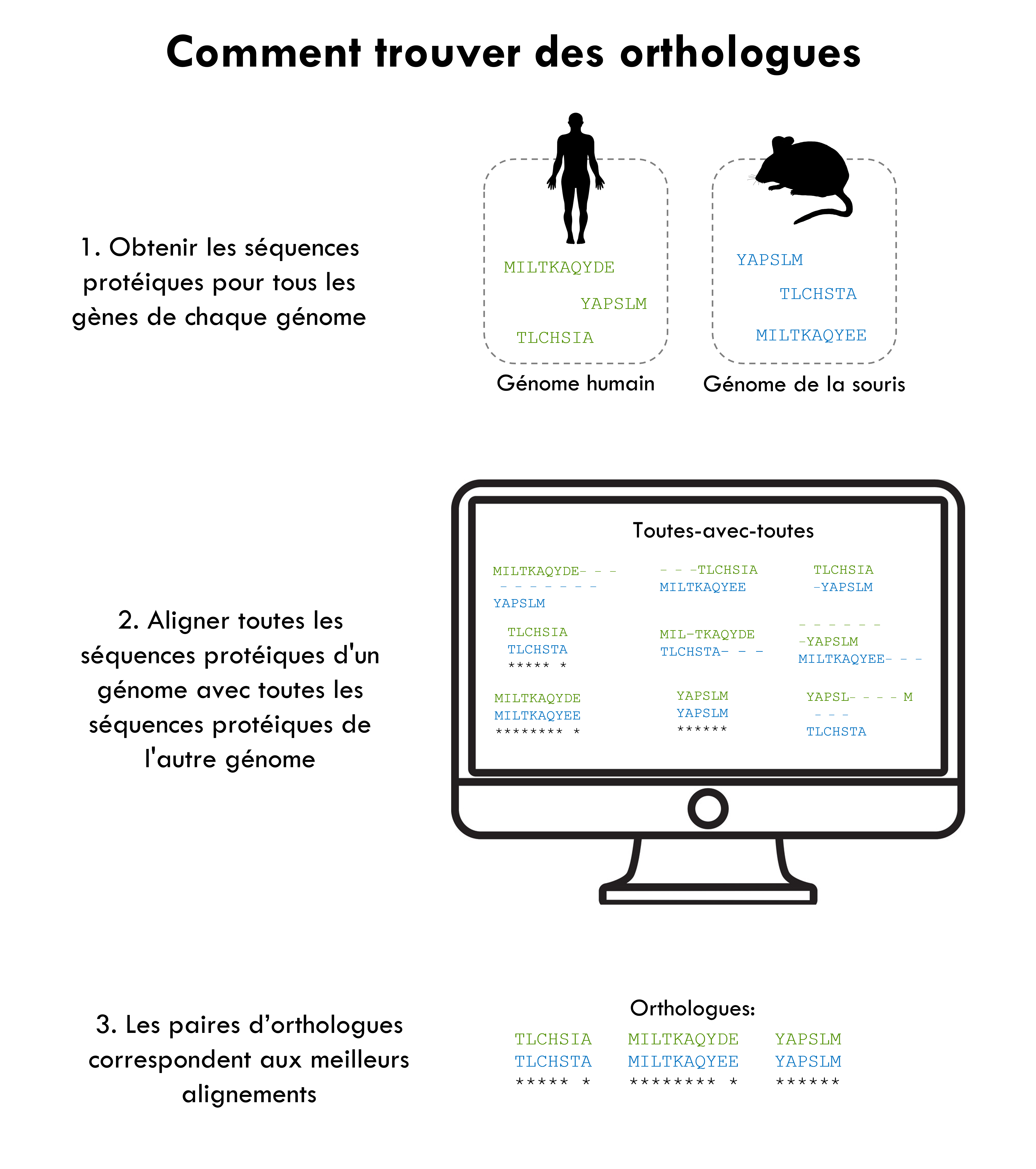
Pour trouver les protéines orthologues, il faut comparer toutes les protéines d’une espèce à toutes les protéines d’une autre espèce afin de trouver lesquelles ‘correspondent’, c’est-à-dire lesquelles sont les plus similaires et donc probablement dérivées d’un gène ancestral commun.
Voici les principales étapes de cette ‘quête des orthologues‘:
1. Récupérer toutes les séquences de protéines produites par un organisme donné.
Ces séquences de protéines sont disponibles dans la banque de données UniProtKB, par exemple. UniProtKB stocke quelque 220 millions de séquences des protéines provenant de plus de 750’000 organismes.
2. Aligner 2 à 2 toutes les protéines d’un organisme avec celles d’un autre organisme. Il y a beaucoup d’alignements à faire! Nous avons besoin d’ordinateurs puissants pour accomplir tous ces calculs.
3. Les orthologues sont dérivés d’un seul et même gène ancestral présent dans l’ancêtre commun des espèces en question: ils sont généralement identifiés comme les paires de protéines présentant le meilleur score d’alignement. L’algorithme prend en compte tous les processus biologiques qui pourraient avoir eu lieu (duplication de gènes, perte de gènes, …).
Voilà, une fois la liste des orthologues établie, nous pouvons commencer à chercher les similitudes et les différences afin de construire des arbres pour étudier l’évolution !
