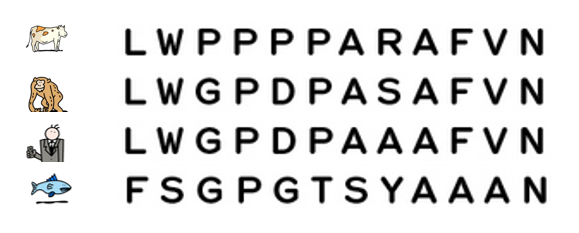Le contexte
10 à 100 millions d’espèces peuplent notre planète.
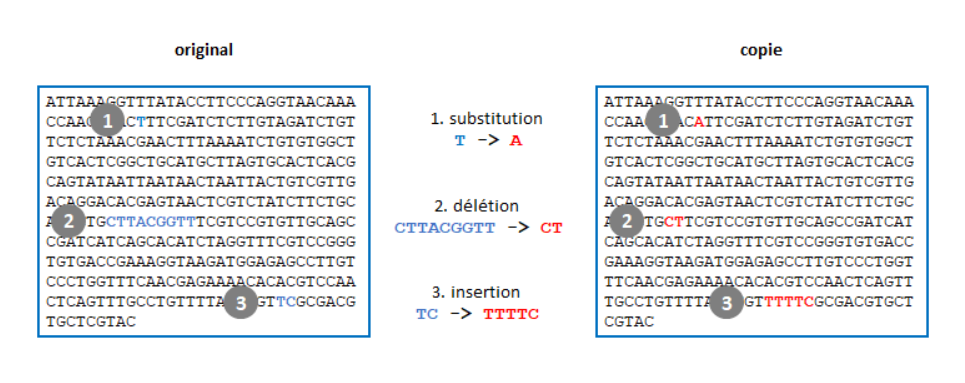
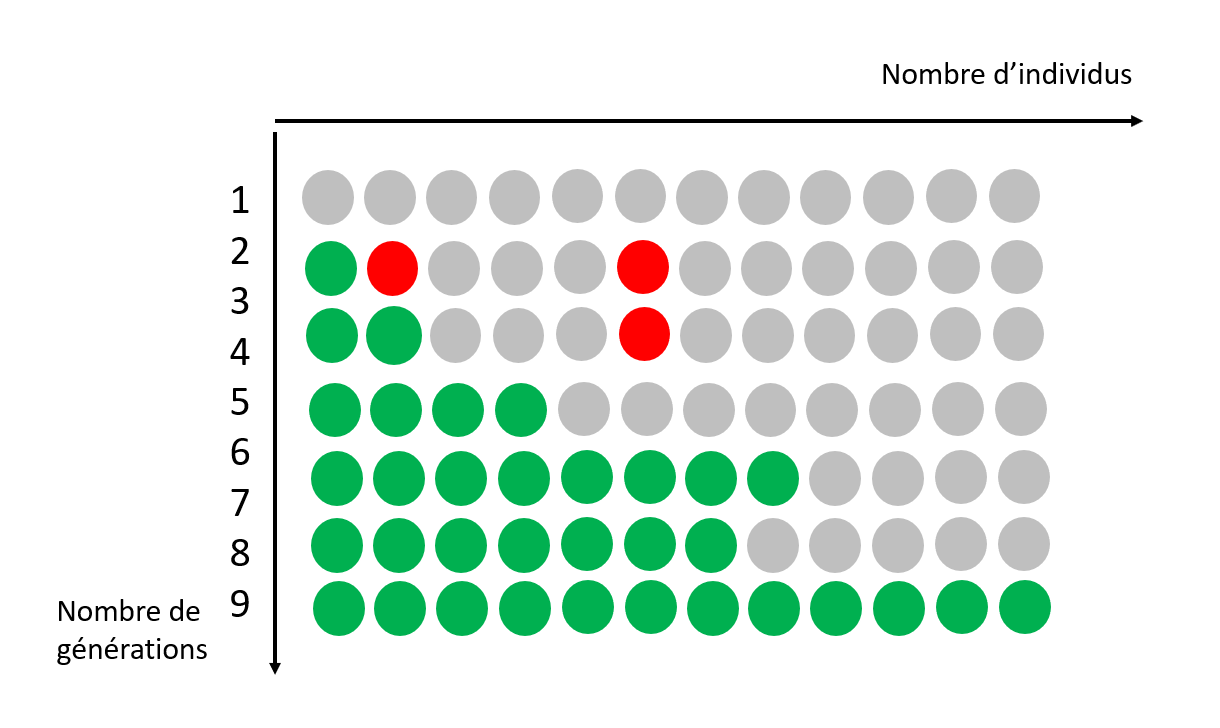
L’évolution est à l’origine de cette diversité: toutes les espèces sont apparentées à des degrés divers. Au sein de chaque espèce, les individus évoluent au fil des générations grâce à des changements qui surviennent de façon aléatoire dans leur ADN.
Ces changements (mutations) vont être sélectionnés, a posteriori, en fonction de l’environnement et de l’interaction des espèces et des individus entre eux. Et parfois, une nouvelle caractéristique voire une nouvelle espèce apparaît, mieux adaptée à son environnement.
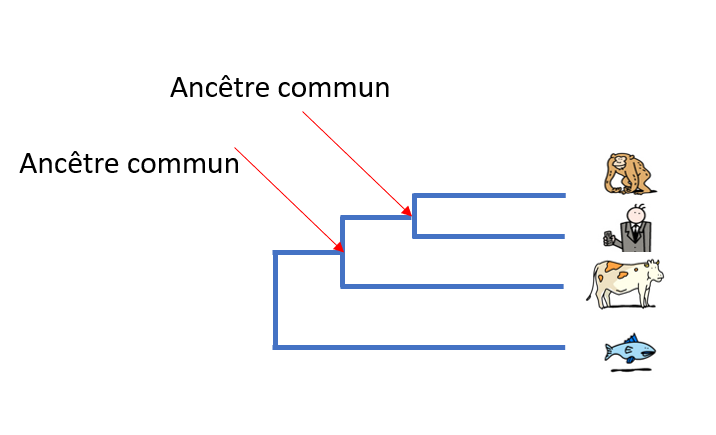
Pour définir les liens de parenté, les biologistes recherchent ce que les espèces ont en commun mais aussi ce qui les distingue. Les liens de parenté entre les différentes espèces sont représentés le plus souvent sous forme d’un arbre, appelé ‘arbre phylogénétique’ ou ‘arbre de la vie’.
A l’époque de Darwin, on commença par comparer les espèces sur la base de leur morphologie – analyses de la taille, de la forme et de la structure des os, la présence de poils ou d’écailles ou, pour les plantes, la position des feuilles sur une tige, par exemple. Plus les caractéristiques morphologiques de 2 espèces sont similaires, plus leur ancêtre commun est récent.
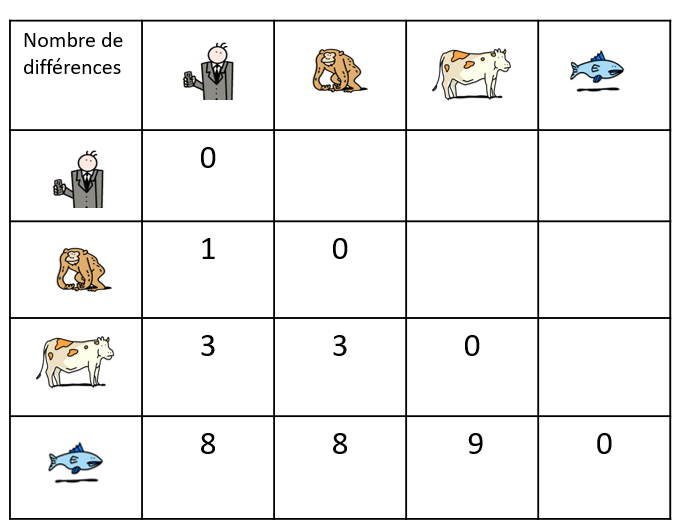
Aujourd’hui, il est possible d’étudier l’évolution des espèces en comparant leur ADN et en particulier, leurs gènes ou leurs protéines. Plus l’ADN de 2 espèces est similaire, plus leur ancêtre commun est récent.