



Ein bisschen Bioinformatik

Hintergrund
10 bis 100 Millionen verschiedene Arten leben auf unserem Planeten.
Die Quelle dieser Vielfalt ist die Evolution: Alle Arten sind in unterschiedlichem Masse miteinander verwandt. Innerhalb jeder Art verändern sich die Individuen über Generationen hinweg durch zufällige Veränderungen in ihrer DNA, sogenannte Mutationen.
Diese Mutationen werden im Zuge der Evolution «selektiert», abhängig von der Umwelt und Wechselwirkungen zwischen Arten und Individuen. Und manchmal taucht ein neues Merkmal oder sogar eine neue Art auf, die besser an ihre Umgebung angepasst ist.
Um den Grad der Verwandtschaft zu bestimmen, untersuchen Biologen, was Arten gemeinsam haben, aber auch, was sie unterscheidet. Die Verwandtschaftsbeziehungen zwischen den verschiedenen Arten werden meistens in Form eines Baumes dargestellt, der als „Stammbaum“ oder „Baum des Lebens“ bezeichnet wird.
Zu Darwins Zeiten begannen die Menschen, Arten anhand ihrer Morphologie zu vergleichen – sie analysierten zum Beispiel Größe, Form und Struktur von Knochen, das Vorhandensein von Haaren oder Schuppen oder bei Pflanzen die Anordnung von Blättern an einem Stängel. Je ähnlicher die morphologischen Merkmale zweier Arten sind, desto jünger ist ihr letzter gemeinsamer Vorfahre.
Heute ist es möglich, die Evolution von Arten zu studieren, indem man ihre DNA und insbesondere ihre Gene oder ihre Proteine vergleicht. Je ähnlicher die DNA zweier Arten ist, desto jünger ist ihr letzter gemeinsamer Vorfahre.
Was ist Bioinformatik?
Was ist Bioinformatik? Wie sequenziert man ein Genom? Wie kann man Gene vorhersagen? Wie findet man Orthologe?
Die Konstruktion eines Lebensbaums mit Hilfe von molekularen Daten bringt viele Herausforderungen mit sich: Zuallererst muss man auf vergleichbare Daten zugreifen können, beispielsweise Sätze orthologer Proteinsequenzen. Und diese Daten werden aus dem Genom verschiedener Organismen gewonnen. Hier kommt die Bioinformatik ins Spiel!
Wie sequenziert man ein Genom?
1. Sequenzierung
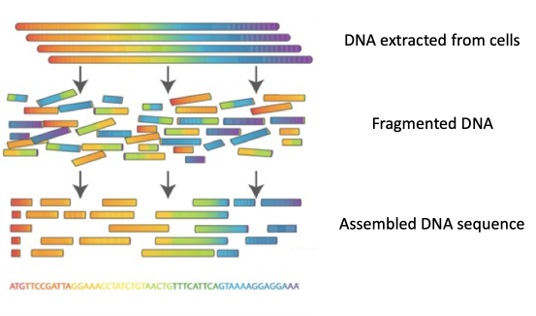
Heutzutage ist es möglich, in wenigen Stunden ganze Genome zu sequenzieren („Whole Genome Sequencing (WGS)“), d.h. die Reihenfolge zu bestimmen, in der die 4 Nukleotide A, C, T, G in einem der DNA-Stränge vorkommen.
Vorgehen: DNA wird aus den Zellen eines Organismus extrahiert und anschließend nach dem Zufallsprinzip in Bruchstücke zerlegt. Jedes Fragment wird mehrere hundert Mal kopiert, bevor es in so genannten DNA-Sequenziergeräten sequenziert wird. Heute können mehr als 10 Milliarden DNA-Fragmente mit einer Länge von jeweils 150 bis 300 Nukleotiden in wenigen Stunden sequenziert werden. Die so erhaltenen DNA-Sequenzen werden dann in einem Computer gespeichert.
Weitere Informationen
Die Revolution der Genomik: neue Sequenzierungsmethoden und ihre Anwendungen
2. Zusammenbauen
Die Milliarden von DNA-Fragmenten (genannt „Reads“) werden dann zusammengesetzt, ein bisschen wie die Teile eines Puzzles, um die Sequenz eines jeden Chromosoms zu rekonstruieren.
Viele Algorithmen sind in der Lage, Genome de novo zusammenzusetzen, also ohne die Hilfe eines Referenzgenoms, das als zuverlässige Vorlage dient – ein bisschen wie ein Puzzle, das ohne Vorlage zusammengesetzt werden müsste.
Jetzt sind Sie dran! DNA-Fragmente zusammensetzen
Viel Spass beim Zusammensetzen dieser 9 auf Papierstreifen gedruckten DNA-Fragmente und dem Finden der DNA-Sequenz des Genoms. Diese DNA-Fragmente überlappen sich gegenseitig und können daher zusammengesetzt werden.
>sequence1 tggtctagctacagtgaaatctcgatgg >sequence2 agctacagtgaaatctcgatgg >sequence3 ggtgattttggtctag >sequence4 ggtgattttggtctagctacagtg >sequence5 ggtgattttggtctagctacag >sequence6 tacagtgaaatctcgatgg >sequence7 ggtgattttggtctagctacagt >sequence8 ttttggtctagctacagtgaaatctcgatgg >sequence9 ggtgattttggtctagctacagt

3. Annotation
Sobald die Genomsequenz bekannt ist, besteht der nächste Schritt darin, herauszufinden, wo sich in diesem riesigen Text die Gene befinden … und darin, ihre biologischen Funktionen vorherzusagen. Dies wird als „Annotation des Genoms“ bezeichnet.’.
Das ist nicht einfach …

Wie kann man Gene identifizieren?
Gene haben sehr variable Längen (zwischen 1000 und 10 Millionen Nukleotiden) und sind im Genom verstreut. Die Gene, die beim Menschen für Proteine kodieren, machen nur einen winzigen Teil – weniger als 5 % – unserer DNA aus. Es ist wie die Suche nach der Nadel im Heuhaufen!
Eine Datei, die die Sequenz eines Genoms von 3 Milliarden Nukleotiden enthält (zum Beispiel das menschliche Genom), würde 3 GB Speicherplatz belegen! Um Ihnen eine Vorstellung von der Grössenordnung der Aufgabe zu geben, hier nur die DNA-Sequenz des menschlichen Chromosoms 1 …
Aus diesem Grund wurden Bioinformatik-Programme entwickelt, die in der Lage sind, den Anfang und das Ende eines Gens, die Exons und die Sequenz des entsprechenden Proteins vorherzusagen.
Diese Software sucht unter anderem nach kleinen Nukleotidsequenzen, von denen bekannt ist, dass sie in Genen vorhanden sind (in grün). Diese Programme sind noch weit davon entfernt, perfekte Ergebnisse zu liefern, aber sie bieten Biologen wertvolle Hinweise. Tatsächlich gibt es heute viel zu viele sequenzierte Genome, als dass all diese Daten experimentell validiert werden könnten!

Die Detektivarbeit ist noch nicht vorbei! Bioinformatiker müssen nun noch nach Orthologen (hauptsächlich orthologen Proteinen) suchen, damit sie anfangen können, sie zu vergleichen, um Ähnlichkeiten und Unterschiede zu untersuchen … und so mehr über die Evolution zu lernen.
Wie findet man Orthologe?
Das menschliche Protein Insulin ist ein Ortholog des gleichnamigen Proteins beim Makaken und beim Rind.
Diese 3 Proteine haben ein gemeinsames Vorfahren-Gen. Woher weiss man das?
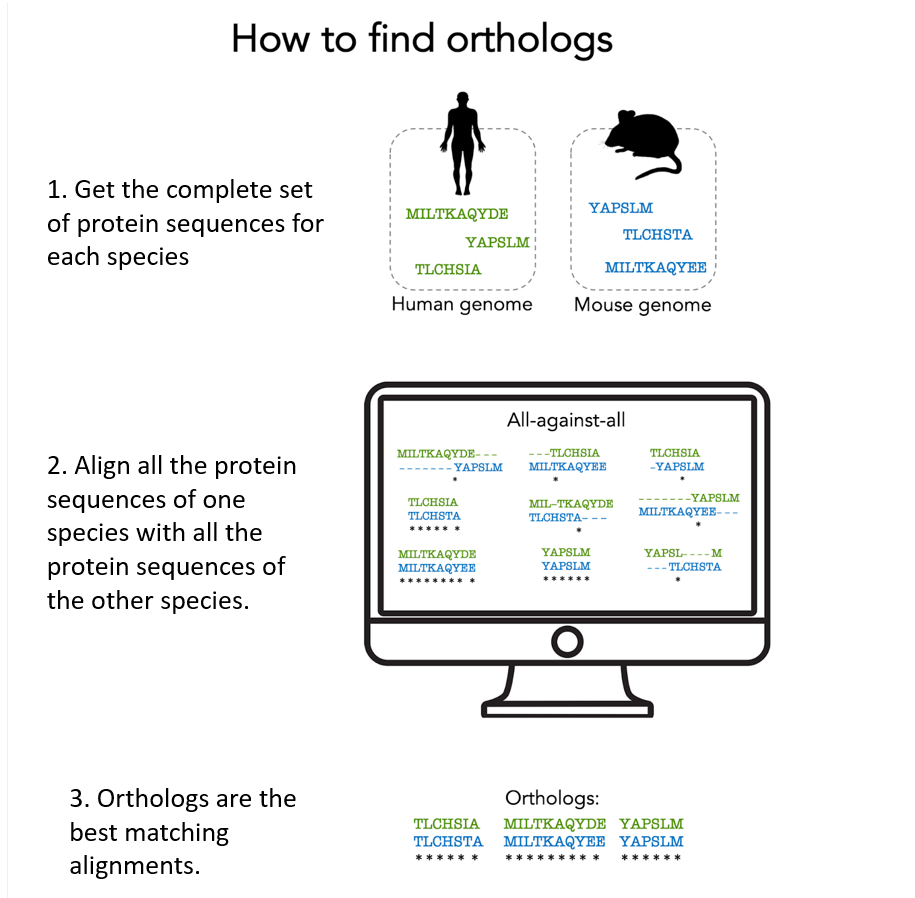
Um orthologe Proteine zu finden, muss man alle Proteine einer Art mit allen Proteinen einer anderen Art vergleichen, um herauszufinden, welche «übereinstimmen», d.h. welche am ähnlichsten sind und daher wahrscheinlich von einem gemeinsamen Vorfahren-Gen stammen.
Hier sind die wichtigsten Schritte dieser „Suche nach Orthologen“:
1. Abruf aller Proteinsequenzen, die von einem bestimmten Organismus produziert werden.
Diese Proteinsequenzen sind beispielsweise in der UniProtKB-Datenbank verfügbar. UniProtKB speichert etwa 230 Millionen Proteinsequenzen von mehr als 750.000 Organismen.
2. Paarweiser Abgleich aller Proteine eines Organismus (einer Art) mit denen eines anderen Organismus (d.h. ein sogenanntes Alignment machen). Das ergibt sehr viele Alignments! Wir brauchen leistungsfähige Computer, um all diese Berechnungen durchzuführen.
3. Orthologe leiten sich von ein und demselben Vorfahren-Gen ab, das beim gemeinsamen Vorfahren der betreffenden Arten zu finden ist: Sie werden im Allgemeinen dadurch identifizert, dass sie die Proteinpaare mit dem besten Alignment-Score sind. Der Algorithmus berücksichtigt alle biologischen Prozesse, die stattgefunden haben könnten (Genduplikation, Genverlust usw.).
Sobald die Liste der Orthologen erstellt ist, können wir hier nach Ähnlichkeiten und Unterschieden suchen, um Bäume zum Studium der Evolution zu erstellen!
