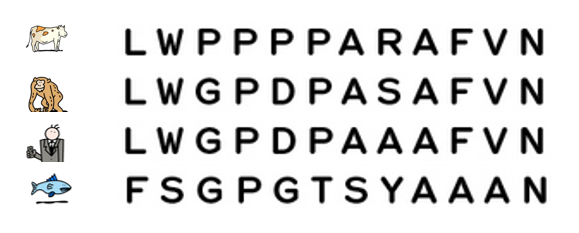Hintergrund
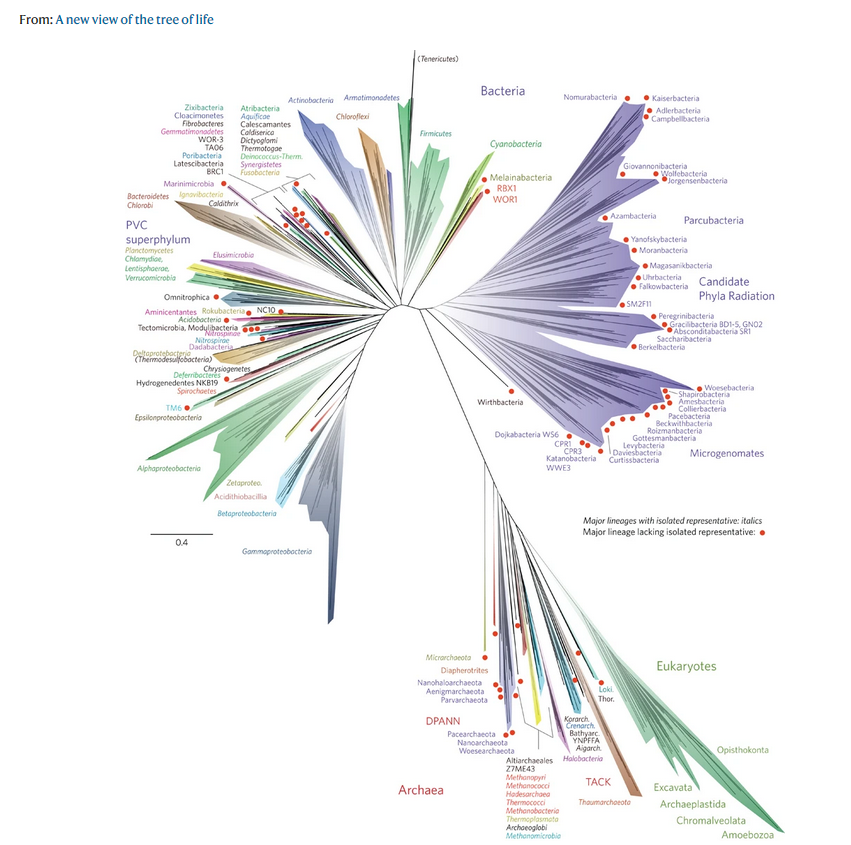
10 bis 100 Millionen verschiedene Arten leben auf unserem Planeten.
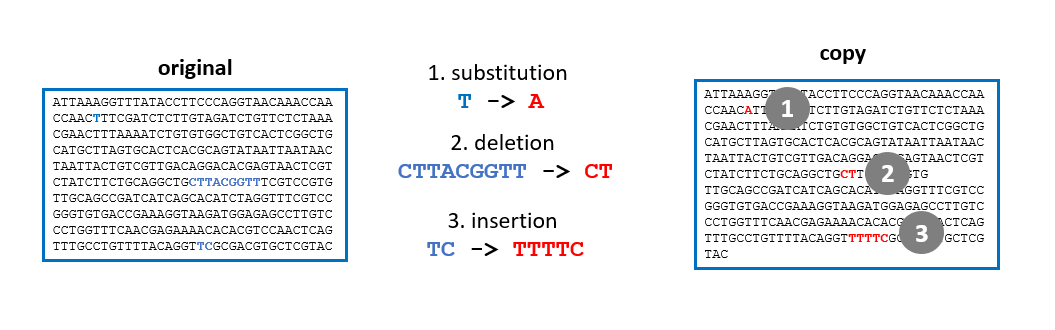
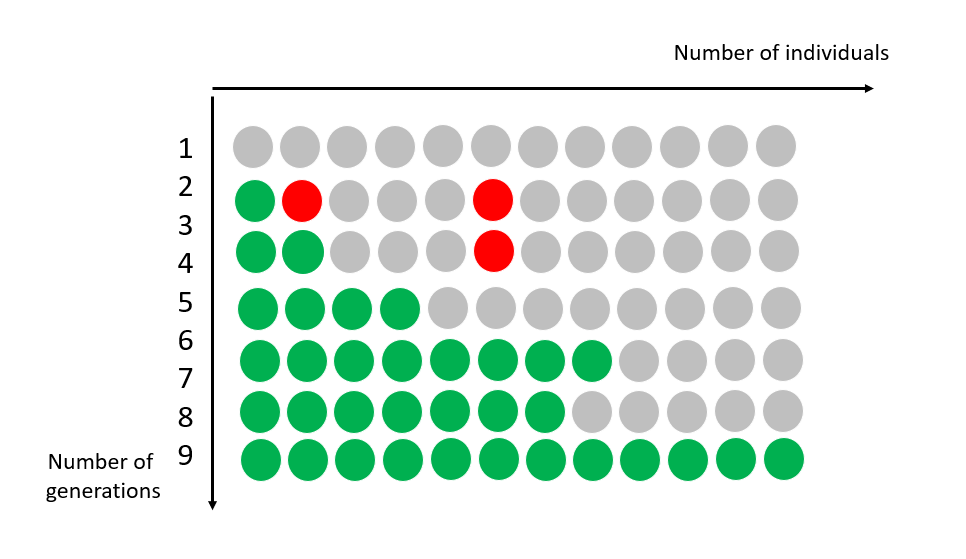
Die Quelle dieser Vielfalt ist die Evolution: Alle Arten sind in unterschiedlichem Masse miteinander verwandt. Innerhalb jeder Art verändern sich die Individuen über Generationen hinweg durch zufällige Veränderungen in ihrer DNA, sogenannte Mutationen.
Diese Mutationen werden im Zuge der Evolution «selektiert», abhängig von der Umwelt und Wechselwirkungen zwischen Arten und Individuen. Und manchmal taucht ein neues Merkmal oder sogar eine neue Art auf, die besser an ihre Umgebung angepasst ist.
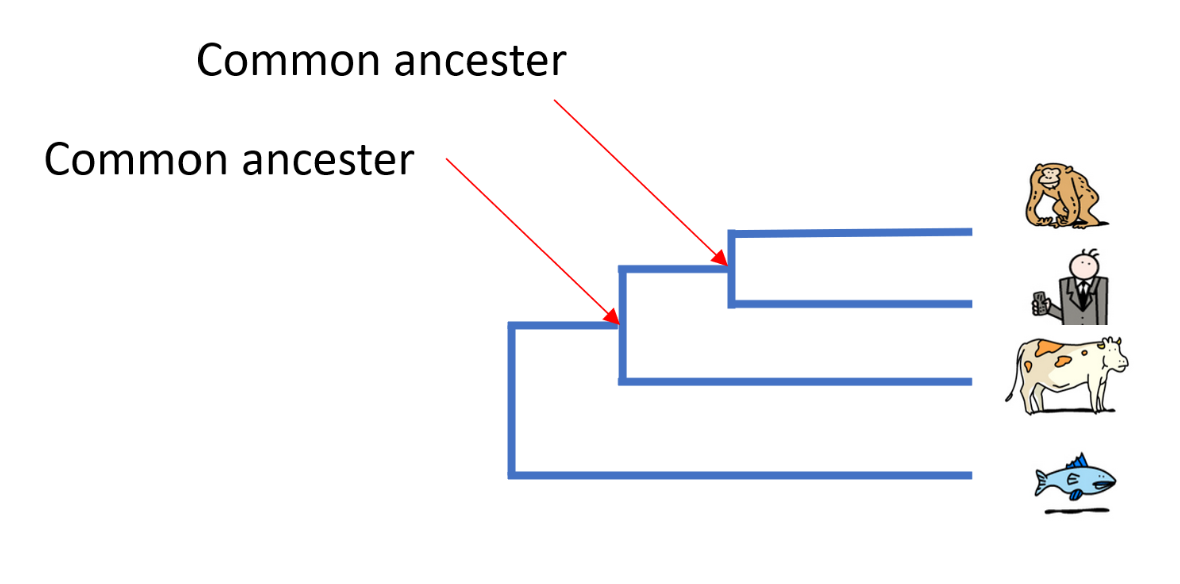
Um den Grad der Verwandtschaft zu bestimmen, untersuchen Biologen, was Arten gemeinsam haben, aber auch, was sie unterscheidet. Die Verwandtschaftsbeziehungen zwischen den verschiedenen Arten werden meistens in Form eines Baumes dargestellt, der als „Stammbaum“ oder „Baum des Lebens“ bezeichnet wird.
Zu Darwins Zeiten begannen die Menschen, Arten anhand ihrer Morphologie zu vergleichen – sie analysierten zum Beispiel Grösse, Form und Struktur von Knochen, das Vorhandensein von Haaren oder Schuppen oder bei Pflanzen die Anordnung von Blättern an einem Stängel. Je ähnlicher die morphologischen Merkmale zweier Arten sind, desto jünger ist ihr letzter gemeinsamer Vorfahre.
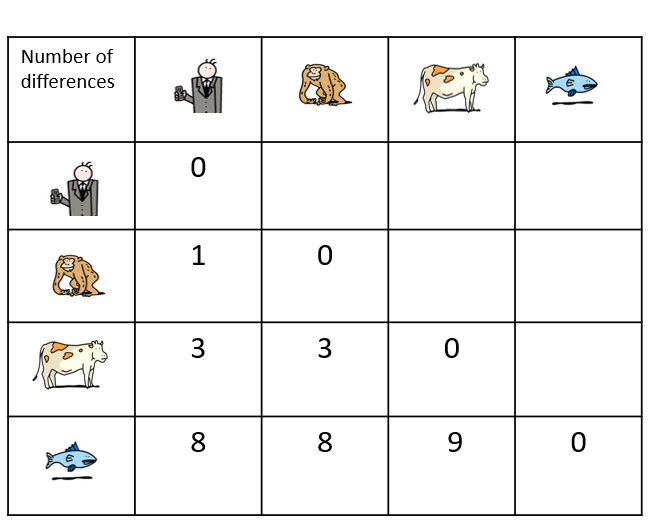
Heute ist es möglich, die Evolution von Arten zu studieren, indem man ihre DNA und insbesondere ihre Gene oder ihre Proteine vergleicht. Je ähnlicher die DNA zweier Arten ist, desto jünger ist ihr letzter gemeinsamer Vorfahre.